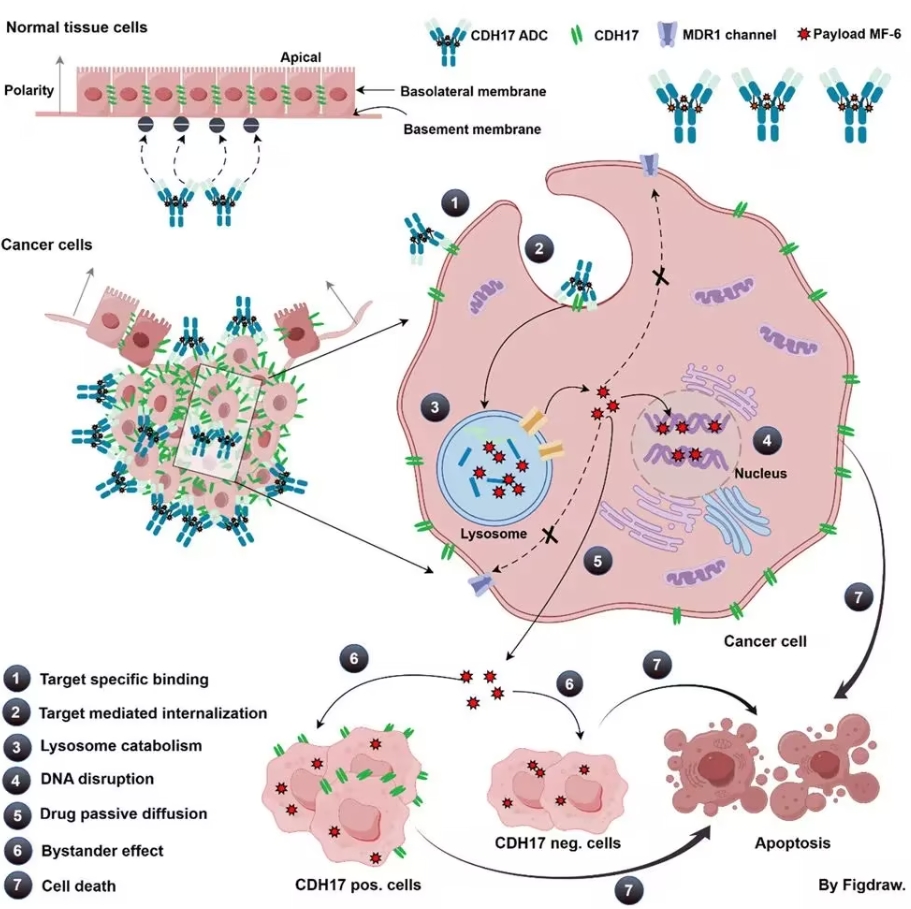
近日,中国首款获批上市的干细胞药物——艾米迈托赛注射液(商品名:睿铂生)在北京大学人民医院开出第一张处方,用于单倍体移植患者的治疗。该药物的单次费用为19800元,一个完整疗程约需8次,总费用为15.8万元。与国际同类产品相比,艾米迈托赛展现出了显著的性价比优势,也为中国患者带来了更多希望。 首个获批干细胞药物:瞄准急性移植物抗宿主病 艾米迈托赛注射液由铂生卓越生物科技(北京)有限公司开发,于2025年1月2日通过国家药监局优先审评程序获批,成为我国首个上市的间充质干细胞(MSC)治疗药物。其适应症为14岁以上患者中以消化道受累为主的、激素治疗失败的急性移植物抗宿主病(aGVHD)。 间充质干细胞因其来源广泛、免疫原性低以及多向分化能力强等特性,成为全球干细胞疗法研究的核心。目前全球已有超过1300项MSCs相关临床试验,涵盖骨科、心血管和免疫性疾病等多个领域。艾米迈托赛作为中国第一款正式获批的MSC药物,标志着我国干细胞技术从研发走向了实际应用。 国际定价对比:突破“天价”壁垒 与国际市场相比,艾米迈托赛注射液治疗总成本展现了显著的成本控制优势。例如,美国FDA在2024年底批准的间充质干细胞药物Ryoncil,用于治疗2个月以上儿童患者的类固醇难治性aGVHD,其单次费用高达19.4万美元(约139万元人民币),一个疗程的费用高达1114万元人民币,是艾米迈托赛疗程费用的70倍。 日韩作为干细胞药物的先行国家,其产品价格也位居高位: 韩国Cartistem(退行性关节炎):每剂1.9万至2.1万美元; 日本Temcell(治疗aGVHD的骨髓间充质干细胞):每袋7700美元,一个标准疗程需8袋,总费用高达12.3万至18.5万美元。 相比之下,艾米迈托赛注射液的定价仅为单剂19800元人民币,为患者节省了大部分治疗成本,展现了“中国智造”在成本管控上的优势。 技术创新:规模化与高效率生产的核心驱动 艾米迈托赛注射液采用了创新性的脐带来源间充质干细胞,这种来源不仅易获取,还特别适合大规模生产。此外,该药物的生产工艺应用了清华大学生物医学工程学院的三维微载体干细胞培养技术: 生产效率提升: 单批次即可生产百亿至千亿量级细胞产品,大幅提高生产能力; 成本显著降低: 通过自动化、连续性、密闭式的生产工艺,减少人工操作和生产空间需求,优化了供应链环节。 这种技术突破有效降低了干细胞生产的难度和成本,为中国干细胞疗法大规模普惠化提供了有力支持。 市场环境与政策推动 近年来,中国干细胞治疗相关政策的频密出台为该领域注入了强劲动力: 2024年初: 《间充质干细胞防治移植物抗宿主病临床试验技术指导原则》发布,为干细胞药物研发提供了专业指导; 2024年5月: 北京药监局核发全国首张干细胞《药物生产许可证》; 2025年6月: 《先进治疗药品的范围、归类和释义(征求意见稿)》明确了细胞治疗药物的分类,进一步规范了市场准入机制。 截至目前,全国已有逾20款干细胞药物进入临床试验阶段,适应症涵盖GVHD、膝骨关节炎、心力衰竭等常见及罕见疾病。艾米迈托赛作为市场化的成功案例,为后续干细胞药物的开发及落地树立了标杆。 下一个挑战:价格可及性与医保覆盖 尽管艾米迈托赛疗程费用在国际范围内较为亲民,但15.8万元的开销仍超出许多中国家庭的承受能力。目前,高昂费用仍是干细胞疗法普及的障碍之一,尤其是在大部分适应症多为罕见病的背景下,医保谈判和覆盖面临较大阻力。 未来,可探索以下路径提升患者可及性: 推动医保支持: 像艾米迈托赛这样的创新药物,可寻求纳入国家医保目录,借助政策降低患者经济负担。 引入商业保险: 探索“商保+分期支付”等创新模式,为更多患者分担高额治疗费用。 持续技术降本: 通过优化细胞生产工艺,进一步降低制造成本,为未来进一步降低市场价格提供可能性。 结语:干细胞产业的普惠化新起点 艾米迈托赛注射液以全球同类产品难以匹敌的价格优势进入市场,不仅是中国干细胞产业发展的重要里程碑,也体现了“创新驱动+成本控制”的综合实力。随着技术的不断成熟、政策的进一步支持及商业模式的创新,干细胞疗法将逐步释放更多市场潜力,为更多患者带来更经济、高效的治疗选择。 以15.8万元为起点,干细胞治疗的未来将更加普及和多元,为医药产业的发展开辟了一个以“可及性”为核心的新时代。