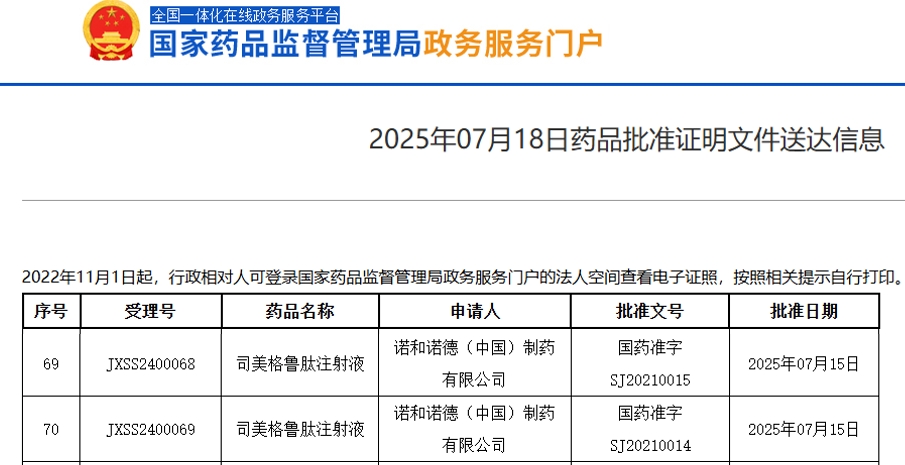
2025年7月14日,武田制药宣布,旗下在研药物**Oveporexton(TAK-861)在治疗1型发作性睡病(NT1)的两项关键III期临床研究中取得显著的正面结果。该药物是一种针对食欲素2型受体(OX2R)**的选择性激动剂,具有潜力成为治疗该疾病的首个同类创新药物。 Oveporexton的临床研究成果 在近期的FirstLight(TAK-861-3001)和RadiantLight(TAK-861-3002)临床试验中,Oveporexton在日间过度嗜睡、猝倒、注意力集中等主要症状的改善上都取得了具有统计学意义的成功。这些研究是在19个国家的多个中心同时进行的,涉及了大规模的患者群体。结果显示,Oveporexton相较于安慰剂组,在第12周时取得了显著的疗效改进,所有主要和次要终点均达成了预期目标,p值均<0.001。 临床疗效全面提升患者生活质量 在这两项研究中,Oveporexton不仅改善了患者的觉醒和日间过度思睡症状,还显著提高了患者的日常生活功能和整体生活质量。研究还表明,服用Oveporexton的患者在猝倒频率、注意力保持等方面都有显著的临床改善,且多项症状的临床指标接近正常范围,展现了该药物在1型发作性睡病治疗中的巨大潜力。 安全性和耐受性良好 Oveporexton在III期研究中的安全性良好,未发现与药物相关的严重不良事件。常见的不良反应包括失眠、尿频和尿急,但总体耐受性较好。研究结束后,超过95%的患者参与了正在进行的长期扩展研究,表明大部分患者对治疗方案保持积极态度。 武田制药的研发战略和未来计划 武田制药在食欲素研究领域持续发力,致力于通过多元化的食欲素产品管线改变治疗现状。Christophe
Weber,武田制药总裁兼CEO表示:“Oveporexton研发的成功标志着我们在治疗难治性疾病方面的显著进步,特别是在1型发作性睡病这一领域,我们有望提供创新的治疗选择。” 武田制药计划在即将召开的医学会议上展示该研究的详细结果,并计划在2025财年向**美国食品药品监督管理局(FDA)**以及全球其他监管机构提交新药申请。 1型发作性睡病的挑战与现有治疗 1型发作性睡病(NT1)是一种极为罕见的神经系统疾病,患者通常会出现日间过度嗜睡、猝倒、夜间睡眠中断、睡眠瘫痪等症状。其发病机制与大脑中的食欲素神经元丢失密切相关,这些神经元对于调节觉醒和睡眠至关重要。当前的治疗手段主要以对症治疗为主,往往只能缓解部分症状,难以全面改善患者的生活质量。 Oveporexton的独特机制和临床优势 Oveporexton作为一种选择性OX2R激动剂,通过刺激食欲素2型受体(OX2R),帮助恢复食欲素信号传导,从而改善因食欲素缺乏导致的症状。该药物通过促进觉醒、减少异常的快速眼动(REM)睡眠现象,显著减轻了NT1患者的日间嗜睡和猝倒症状,为该疾病的治疗提供了新的方向。 武田制药的未来展望 除了Oveporexton,武田制药还在研发多款食欲素产品,其中包括TAK-360,一种口服OX2R激动剂,计划用于治疗2型发作性睡病(NT2)、特发性嗜睡症(IH)等疾病。武田制药的研发战略将继续推动食欲素科学领域的创新,力争为全球患者提供更好的治疗方案。