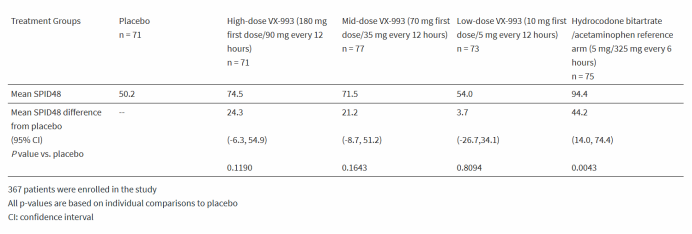
肝细胞癌(Hepatocellular Carcinoma,
HCC)是全球癌症相关死亡的主要原因之一。由于多数患者在晚期确诊,全身系统性治疗成为主要手段。然而,HCC的抗药性问题限制了疗效并导致预后不佳。 2025年8月1日,清华大学董家鸿院士、王韫芳研究员等在《Signal Transduction and Targeted
Therapy》上发表了题为《Targeting AKR1B1 inhibits metabolic reprogramming to reverse
systemic therapy resistance in hepatocellular
carcinoma》的研究论文。这项研究揭示了代谢重编程如何导致HCC对全身系统性治疗的耐药性,并将关键调控因子AKR1B1确定为新的治疗靶点。 AKR1B1:治疗耐药的关键调控酶 研究背景 HCC的多药耐药性为临床治疗带来了巨大挑战。研究发现,肿瘤细胞的代谢重编程是导致耐药的重要机制,但其分子调控网络尚未明确。 核心发现 通过建立HCC多药耐药细胞系,研究团队发现这些细胞显示出葡萄糖-脂质代谢和谷胱甘肽代谢通路的异常活跃,这些代谢途径在肿瘤细胞增殖和存活中发挥重要作用。其中,AKR1B1是代谢重编程的关键调控酶,通过调节肿瘤能量代谢和增强应激耐受性,在耐药性中起核心作用。 研究进一步揭示,AKR1B1表达水平升高与肝细胞癌患者耐药性及不良预后密切相关。此外,AKR1B1的分泌特性促进了耐药性的细胞间传递,突显了其对预测耐药性的临床价值。 靶向AKR1B1的治疗潜力 抑制AKR1B1逆转耐药性 研究创新性地尝试将依帕司他(Epalrestat)——一种已经获批用于治疗糖尿病周围神经病变的AKR1B1抑制剂,联合标准全身治疗药物仑伐替尼应用于HCC治疗。结果显示,这种联合疗法能够显著减轻HCC的耐药性,有效增强标准疗法的抗肿瘤效果。 关键实验数据 代谢评估:通过多组学分析,AKR1B1对HCC耐药细胞代谢重编程的影响得以验证; 临床关联性:患者样本进一步证明了AKR1B1高表达与耐药及预后不良密切相关; 安全与有效性:依帕司他作为临床已上市药物,具备良好的安全性,显著降低HCC耐药性的同时也增强了标准药物的疗效。 代谢重编程与HCC耐药性 肿瘤代谢的再编程 代谢重编程是肿瘤适应恶劣环境的重要手段。HCC耐药细胞表现出: 葡萄糖-脂质代谢增加:为肿瘤增殖提供能量来源; 谷胱甘肽代谢增强:提高细胞对氧化应激的耐受性。 AKR1B1的核心作用 AKR1B1作为一种调控酶,贯穿了上述代谢通路: 增强代谢活动:推动肿瘤细胞增殖和生存; 细胞间传递:通过分泌特性扩散耐药性,从而使肿瘤群体产生集体抗药性。 临床意义:全新视角与治疗策略 提供新的耐药标志物 AKR1B1不仅是肝细胞癌耐药的重要调控因子,其分泌特性还使其成为预测患者耐药风险和治疗效果的潜在生物标志物。 开发联合治疗方案 靶向AKR1B1的联合疗法,如依帕司他与仑伐替尼的组合,不仅在抑制耐药性上表现出显著效果,而且能实际改善患者预后。这为HCC的耐药治疗提供了全新方向。 推动精准医疗 基于AKR1B1的分子机制研究,有望开发出针对特定代谢亚型HCC患者的精准个性化治疗策略,进一步优化治疗效果。 展望:克服HCC耐药性的未来路径 尽管该研究在靶向AKR1B1耐药性上取得突破,仍有一些领域值得进一步研究: 适用人群扩展:验证AKR1B1抑制剂疗法在不同阶段HCC患者中的疗效; 整合治疗手段:探索AKR1B1抑制剂与其他免疫疗法或化疗药物的协同作用; 机制深入解析:更全面地分析AKR1B1在其他代谢通路中的潜在角色,以实现全链条靶向干预。 总结 通过揭示代谢重编程对肝细胞癌耐药性的核心作用,本文研究明确了AKR1B1作为关键调控酶的中心地位,并验证了其靶向阻断耐药途径的可行性。依帕司他与标准疗法的联合应用为克服HCC耐药性提供了重要的治疗思路,为患者带来了新的生存希望。这一突破性发现为肝癌的精准治疗和抗耐药策略发展奠定了坚实基础,未来仍需通过临床研究进一步验证其广泛应用的潜力。